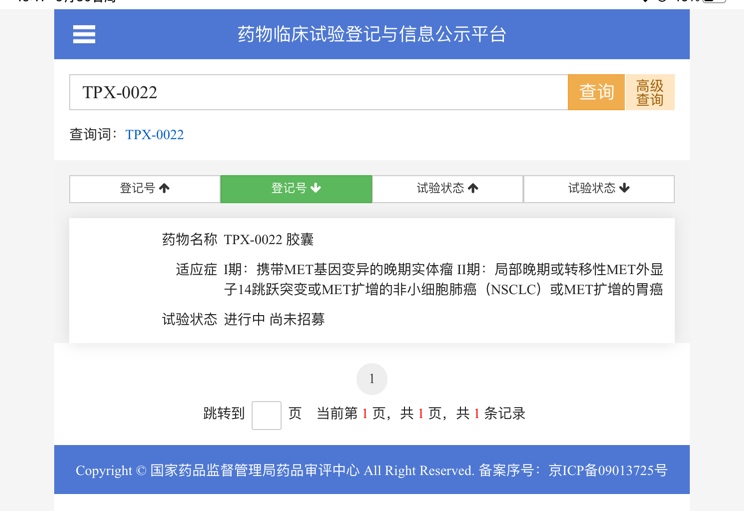
| |
 EGFR靶点病人,到底适不适合免疫治疗
一直一直在听说,肺癌EGFR突变的病人,免疫治疗很难获益,甚至有超进展可能,具体哪里
EGFR靶点病人,到底适不适合免疫治疗
一直一直在听说,肺癌EGFR突变的病人,免疫治疗很难获益,甚至有超进展可能,具体哪里
 惋惜 关于一位术后2期肺腺癌病友止步
曾经一起抗癌的老病友很多都陆陆续续地抗癌结束了,如今都各自有了新的生活,当
惋惜 关于一位术后2期肺腺癌病友止步
曾经一起抗癌的老病友很多都陆陆续续地抗癌结束了,如今都各自有了新的生活,当
 多方聚力,星火成炬 | 医企患同心共
2025年11月8日,在第八届中国国际进口博览会拜耳展台,一场以“多方聚力,星火成炬 |
多方聚力,星火成炬 | 医企患同心共
2025年11月8日,在第八届中国国际进口博览会拜耳展台,一场以“多方聚力,星火成炬 |
 肺鳞癌一线治疗新突破,无进展生存期
作者:seacat
最近举行的欧洲肿瘤内科学会2025年会(ESMO 2025)上,上海交通大学附属
肺鳞癌一线治疗新突破,无进展生存期
作者:seacat
最近举行的欧洲肿瘤内科学会2025年会(ESMO 2025)上,上海交通大学附属
 对话国际肺癌专家,解锁“早筛早治”
作者:雨过天晴肺癌,长期以来都是全球范围内发病率和死亡率最高的癌症之一,成为威胁
对话国际肺癌专家,解锁“早筛早治”
作者:雨过天晴肺癌,长期以来都是全球范围内发病率和死亡率最高的癌症之一,成为威胁





 显身卡
显身卡